
The Overlooked Connection: Dysbiosis and Degenerative Disease
Degenerative musculoskeletal diseases, including osteoarthritis (OA), osteoporosis (OP), sarcopenia (SP), and intervertebral disc degeneration (IVDD), affect well over a billion people globally and share three core pathological features: chronic inflammation, oxidative stress, and dysregulated cell death. What is increasingly clear, and clinically underappreciated, is that gut microbiota (GM) dysbiosis plays a central upstream role in driving all four conditions simultaneously.
A recent article, "Glucagon-like peptide-1: a critical link between gut microbiota dysbiosis and degenerative musculoskeletal diseases", caught my attention for its thoroughness and specificity related to our ongoing discussions and exploration of the pleiotropic power of GLP-1 RAs. In their study, the authors present a framework that looks at the occurrence of dysbiosis in aging populations and how it is characterized by reduced SCFA production, depletion of beneficial bile acids, and increased translocation of lipopolysaccharide (LPS). It's this combination that systematically impairs GLP-1 signaling and thereby accelerates bone loss, muscle atrophy, cartilage degradation, and disc degeneration through shared inflammatory and oxidative pathways.
The authors examine how the mechanism of dysbiosis runs through GLP-1. Intestinal L cells, which are concentrated in the distal ileum and colon where bacterial density peaks, depend heavily on microbial metabolites to trigger GLP-1 secretion. Unlike proximal L cells stimulated by direct nutrient contact, these distal cells rely on short-chain fatty acids (SCFAs), secondary bile acids (BAs), and tryptophan-derived metabolites produced by commensal bacteria. When the gut ecosystem is disrupted by aging or chronic disease, this metabolite-driven GLP-1 stimulation fails. The result is a state of GLP-1 deficiency that is not primarily dietary in origin and therefore cannot be corrected by diet alone. The study then confirmed that depleting gut bacteria via antibiotics or germ-free mouse models essentially abolishes postprandial GLP-1, while fecal microbiota transplantation (FMT) restores it.
Microbial Pathways Regulating GLP-1
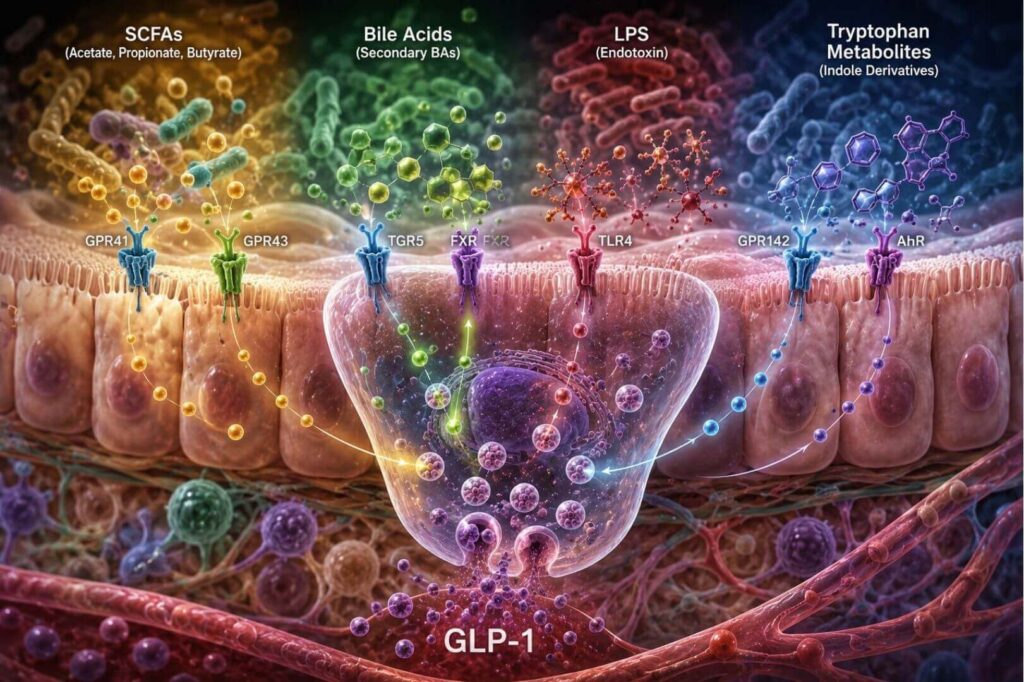
As I’ve discussed in other articles, as well as in my book, The Quantum Power of GLP-1 Peptides, the authors of this study frame their exploration by first establishing the four principal microbial metabolite pathways that drive GLP-1 secretion from intestinal L cells, including
- SCFAs (acetate, propionate, butyrate) produced by bacteria such as Faecalibacterium prausnitzii, Roseburia intestinalis, Akkermansia muciniphila, and Bifidobacterium activate GPR41 and GPR43 receptors on L cells, triggering calcium influx and GLP-1 exocytosis. Propionate shows particularly high efficacy, and variations in the Firmicutes-to-Bacteroidetes ratio may meaningfully shape incretin responses independently of total SCFA levels.
- Bile acids, transformed from primary to secondary forms by bacteria including Clostridium bolteae and Bacteroides, activate TGR5 receptors and antagonize FXR signaling on L cells, both of which promote GLP-1 synthesis and release. GUDCA and UDCA derived from C. bolteae have been shown to directly promote proglucagon gene transcription and stimulate intestinal stem cell proliferation, effectively increasing the number of GLP-1–secreting L cells.
- LPS, released by gram-negative bacteria during barrier disruption, exhibits a biphasic effect: at low doses it activates TLR4 on L cells and acutely stimulates GLP-1 release; at high chronic doses it impairs L cell function and drives GLP-1 resistance. This "functional switch" is clinically relevant in that low-grade metabolic endotoxemia common in aging and metabolic syndrome may initially upregulate GLP-1, but sustained dysbiosis ultimately exhausts this response.
- Tryptophan metabolites, particularly indole derivatives produced by Lactobacillus species and Akkermansia muciniphila, promote L cell differentiation and activate GLP-1 secretion via GPR142 and the aryl hydrocarbon receptor.
Four Core Mechanisms of GLP-1 Tissue Protection
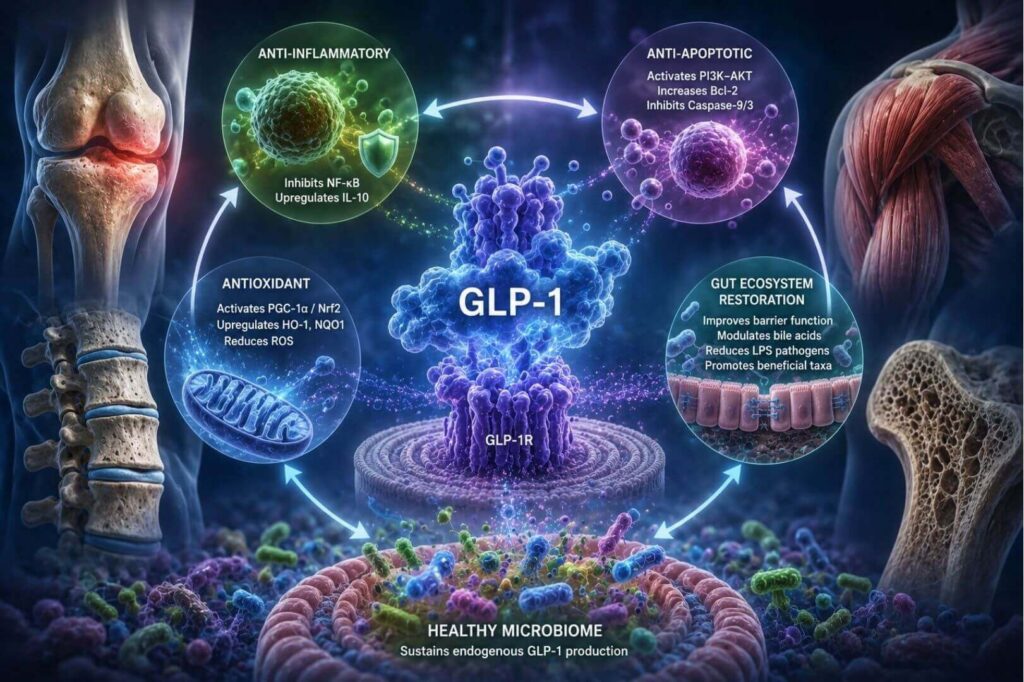
Once secreted, GLP-1 operates through a remarkably conserved set of protective mechanisms across all four musculoskeletal tissues, acting via the ubiquitously expressed GLP-1 receptor (GLP-1R). First, GLP-1 suppresses NF-κB signaling by reducing IκBα phosphorylation, downregulating TNF-α, IL-6, and IL-1β. Simultaneously, it activates the cAMP–PKA–CREB axis to upregulate IL-10. Together these mechanisms deliver an anti-inflammatory effect. The authors note that this relationships has been "validated across multiple disease models, including synovial macrophages in OA, bone marrow precursor cells in OP, and muscle cells in SP."
Additionally, the cellular mechanisms serve an antioxidant purpose via PGC-1α activation and Nrf2 nuclear translocation, GLP-1 upregulates HO-1 and NQO1 while reducing NADPH oxidase activity and ROS generation; together these mechanisms protect mitochondrial integrity across cartilage, bone, muscle, and disc tissue. Further, through PI3K–AKT signaling, GLP-1 suppresses Bax, enhances Bcl-2 expression, and inhibits Caspase-9/3 activation, preserving cell viability under degenerative stress – offering an anti-apoptotic effect.
Finally, GLP-1 itself acts reciprocally on the gut ecosystem by enhancing tight junction integrity, modulating bile acid metabolism, suppressing LPS-producing pathogens, and promoting colonization by beneficial taxa including Akkermansia muciniphila and Roseburia. This bidirectional loop means GLP-1 receptor agonists (GLP-1RAs) do not merely supplement a deficient hormone; they help restore the microbial environment that sustains endogenous GLP-1 production.
A Deeper Look at the Cellular Mechanisms and Signaling Pathways

At the level of intestinal L cells, GLP-1 secretion is orchestrated through a sophisticated array of receptor-coupled signaling cascades. SCFA-producing bacteria generate propionate and butyrate that bind to free fatty acid receptors FFAR2 (GPR43) and FFAR3 (GPR41) on the L cell surface. FFAR2, being Gq-coupled, triggers an influx of intracellular calcium ions (Ca²⁺) that directly drives GLP-1 exocytosis — an effect that disappears entirely in FFAR2-deficient models. Simultaneously, SCFA metabolism generates ATP, which causes membrane depolarization and a secondary Ca²⁺ influx, further amplifying secretion. MAPK signaling (via p-ERK and p-p38) upregulates both FFAR and GLP-1R expression, while the farnesoid X receptor (FXR) acts as a brake on this system and thereby suppress FFAR2 expression and dampen SCFA-induced GLP-1 release when activated. Bile acid pathways add another layer: secondary bile acids produced by gut bacteria bind basolateral TGR5 receptors on L cells, activating the Gαs–cAMP–CREB signaling cascade that raises intracellular cAMP and calcium, promoting GLP-1 granule exocytosis. Conversely, when Clostridium bolteae–derived UDCA and GUDCA act as FXR antagonists, they relieve FXR-mediated repression of the proglucagon gene (Gcg), upregulate glycolytic enzymes phosphofructokinase 1 and enolase 1 to increase ATP production, and additionally stimulate proliferation of Lgr5⁺ intestinal stem cells and effectively expand the pool of GLP-1–secreting L cells rather than merely increasing output per cell. Tryptophan metabolites, including indole-3-lactic acid from Lactobacillus species activate GPR142 on L cells and promote the differentiation of intestinal stem cells toward the L cell lineage by upregulating transcription factors Math1, Neurog3, and Neurod1 while suppressing Notch signaling, making tryptophan metabolism a long-term structural regulator of GLP-1 secretory capacity, not merely an acute trigger.
Once GLP-1 binds its receptor on musculoskeletal target tissues, it activates a convergent but tissue-tuned set of intracellular pathways. The primary signaling axis includes GLP-1R → adenylate cyclase → cAMP → PKA and then branches into three protective programs that ultimately converge on NF-κB suppression. First, PKA phosphorylates CREB, which simultaneously promotes anti-inflammatory IL-10 transcription and directly inhibits NF-κB nuclear translocation, reducing TNF-α, IL-6, and IL-1β production. Second, PKA activates PGC-1α, the master regulator of mitochondrial biogenesis, which stabilizes mitochondrial membrane potential, enhances fatty acid oxidation, and activates Nrf2, a transcription factor that upregulates antioxidant enzymes HO-1 and NQO1 while suppressing NADPH oxidase (NOX) to reduce reactive oxygen species generation. Third, GLP-1 engages the PI3K–AKT–MAPK axis in parallel: AKT suppresses pro-apoptotic Bax while upregulating Bcl-2, preventing mitochondrial outer membrane permeabilization and the downstream caspase-9/caspase-3 cascade that executes apoptosis; MAPK signaling simultaneously reinforces Nrf2 nuclear translocation, creating a redundant antioxidant defense. In osteoblasts specifically, AKT promotes GLUT4 translocation via PI3K signaling to restore glucose uptake under insulin-resistant conditions, securing the carbon substrates needed for collagen glycosylation and ATP-dependent mineralization, a metabolic reprogramming function that extends well beyond the hormone's classical glycemic role. In intervertebral disc cells, GLP-1 adds a further layer of transcriptional specificity: rather than broadly suppressing AP-1, it selectively disrupts the formation of BATF/JUN heterodimers — the principal drivers of MMP13 and ADAMTS5 transcription — while leaving other AP-1 configurations intact. Concurrently, GLP-1 enhances SIRT1 expression, which deacetylates the RelA/p65 subunit of NF-κB to limit its nuclear translocation, suppressing IL-1β and TNF-α at the transcriptional level. In infiltrating macrophages within the disc microenvironment, GLP-1 potentiates STAT3 signaling to promote M2 polarization, fostering a pro-regenerative immune niche, while paradoxically attenuating pathological STAT3 phosphorylation in nucleus pulposus cells themselves to reduce cellular senescence. This cell-type–specific modulation of the same signaling pathway represents one of the more remarkable features of GLP-1's mechanistic repertoire.
The bidirectional nature of the GM–GLP-1 axis adds a self-reinforcing dimension that carries significant clinical implications. GLP-1 and GLP-1RAs do not merely respond to microbial signals: they actively remodel the gut ecosystem in ways that amplify their own therapeutic effects. By strengthening tight junction proteins (occludin, claudin-1, ZO-1), GLP-1 reduces LPS translocation and reshapes the luminal environment to suppress dysbiotic gram-negative overgrowth. By modulating bile acid metabolism through effects on Clostridium bolteae and related species, GLP-1RAs alter the composition of the secondary bile acid pool in ways that further stimulate TGR5 and antagonize FXR, reinforcing GLP-1 secretion from L cells. GLP-1 analog treatment increases colonic indole-3-propionic acid (IPA) levels, which in turn supports L cell function and barrier integrity. At the population level, GLP-1RAs have been shown to enrich SCFA-producing genera including Roseburia and Ruminococcus and promote Akkermansia muciniphila colonization (a bacterium whose bioactive protein P9 independently stimulates further GLP-1 secretion) while simultaneously suppressing pathobionts including Escherichia coli and Enterococcus by reducing endotoxemia and restoring barrier function. The clinical consequence is a positive feedback architecture: pharmacological GLP-1R activation improves the microbial environment, which elevates endogenous GLP-1, which further refines microbial ecology, collectively amplifying anti-inflammatory, osteoanabolic, and myoprotective downstream effects across musculoskeletal tissues simultaneously.
Specific Clinical Relevance
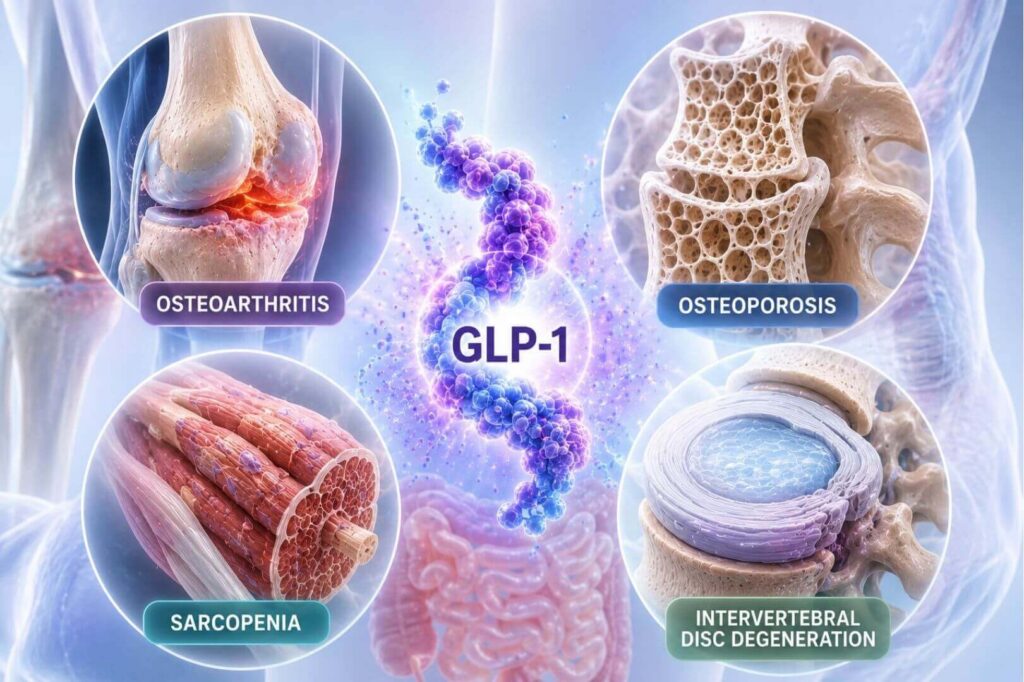
Osteoarthritis
GLP-1's primary advantage in OA is its dual capacity to modulate synovial macrophage polarization (M1→M2) while directly inhibiting cartilage matrix-degrading enzymes (MMP-3, MMP-13, ADAMTS-4/5) and promoting COL2 and aggrecan synthesis. The STEP 9 clinical trial (n=407) demonstrated that weekly semaglutide 2.4 mg over 68 weeks produced greater pain reduction than placebo on WOMAC scores, with an NSAID-sparing effect; improvements exceeded what could be attributed to weight loss alone. A Shanghai cohort study of 1,807 knee OA patients with type 2 diabetes showed GLP-1RA users had slower medial tibiofemoral cartilage loss and a reduced rate of knee surgery.
Osteoporosis
GLP-1 promotes osteoblast proliferation and differentiation while suppressing osteoclastogenesis via NF-κB/MAPK inhibition and OPG/RANKL rebalancing. It also indirectly inhibits bone resorption through calcitonin release from thyroid C-cells. Despite concerns that GLP-1RA–associated weight loss reduces BMD through mechanical unloading (Wolff's law), a 2025 retrospective cohort of elderly type 2 diabetes patients showed long-term GLP-1RA use was associated with a significantly lower incidence of osteoporosis (HR 0.69). A 2024 randomized trial found that combining GLP-1RA therapy with resistance exercise fully preserved BMD at hip and lumbar spine despite greater weight loss than pharmacotherapy alone — a critical practical point for clinical management.
Sarcopenia
GLP-1's mechanism in muscle involves upregulating myogenic transcription factors (MyoD, MyoG) while suppressing atrophy genes (Atrogin-1, MuRF-1, myostatin) and enhancing mitochondrial biogenesis. Importantly, the paper draws a clinically significant distinction: in rodent models, GLP-1R is directly expressed on myocytes, enabling fiber-type transitions; in humans, functional GLP-1R in skeletal muscle is predominantly localized to vascular endothelial and smooth muscle cells. The primary human mechanism is therefore indirect: GLP-1–induced microvascular recruitment increases capillary perfusion and oxygen delivery, alleviating hypoxia-driven muscle dysfunction. The clinical implication is direct: "although patients exhibit improved endurance... absolute muscle strength remains susceptible to decline." Preserving strength requires concurrent resistance training and high-protein intake (>1.2g/kg/day).
Intervertebral Disc Degeneration
GLP-1 protects nucleus pulposus cells (NPCs) ( the primary cell type driving disc integrity) by inhibiting AP-1–driven catabolic gene transcription (specifically BATF/JUN heterodimers that drive MMP13 and ADAMTS5), curtailing NF-κB via SIRT1 upregulation, and differentially modulating STAT3 (suppressing pathological senescence in NPCs while promoting M2 macrophage polarization). Intradiscal exenatide injection in animal models significantly improved nucleus pulposus hydration, proteoglycan content, and disc height on MRI. A retrospective cohort study found GLP-1RA use was associated with progressively lower rates of lumbar spine surgery over five years.
Therapeutic Strategy: Targeting the GM–GLP-1 Axis
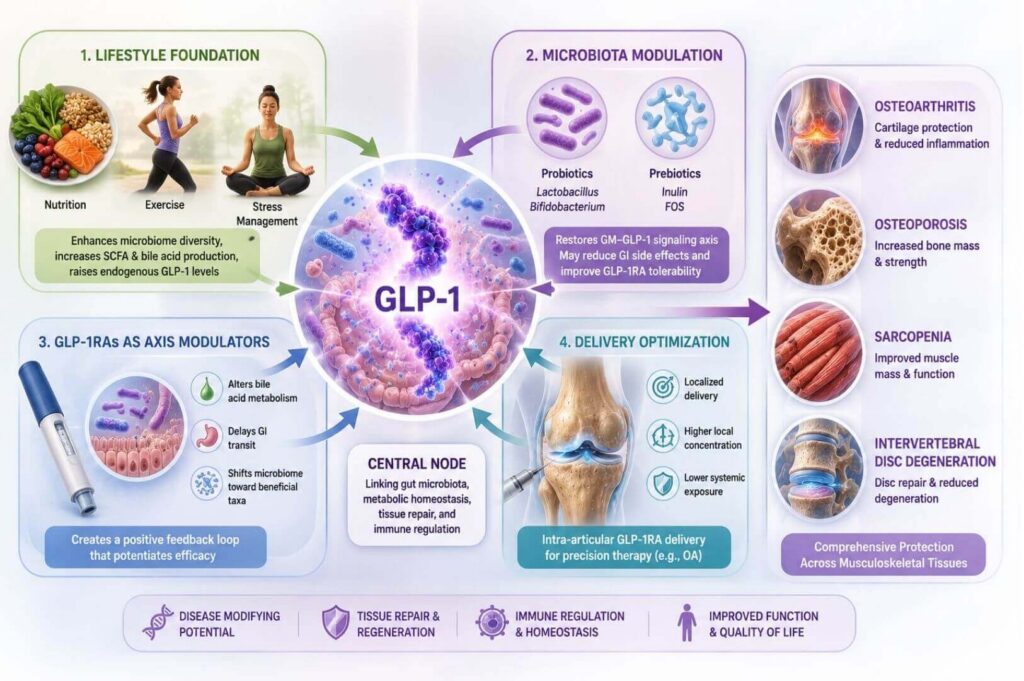
The authors propose a multi-level intervention framework that moves well beyond GLP-1RA pharmacotherapy alone:
- Lifestyle foundation: Diet and exercise modulate GM composition, enhance SCFA and bile acid production, and raise endogenous GLP-1 levels — simultaneously addressing all four disease processes.
- Microbiota modulation: Targeted probiotics (Lactobacillus, Bifidobacterium) and prebiotics (inulin, fructooligosaccharides) restore the GM–GLP-1 signaling axis and may mitigate GLP-1RA gastrointestinal side effects that limit dose escalation. Ongoing trials are investigating probiotic co-administration specifically during GLP-1RA titration.
- GLP-1RAs as axis modulators: Beyond their direct tissue-protective actions, GLP-1RAs alter bile acid metabolism, delay GI transit, and shift microbial ecology toward beneficial taxa, creating a positive feedback loop that potentiates their own efficacy.
- Delivery optimization: For localized conditions like OA, intra-articular GLP-1RA delivery may achieve higher local concentrations with less systemic exposure, representing a precision medicine opportunity.
The authors conclude that GLP-1 functions as "a central node linking gut microbiota, metabolic homeostasis, tissue repair, and immune regulation," a framing that positions GLP-1 pathway intervention not as a metabolic adjunct but as a legitimate disease-modifying strategy across the spectrum of age-related musculoskeletal degeneration.
References
Yang, W., Hao, C., Wang, N., Xie, J., Zhou, B., Yang, Z., Zheng, A., Wei, J., Li, C., Xie, C., Li, H., Lei, G., & Zeng, C. (2026). Glucagon-like peptide-1: a critical link between gut microbiota dysbiosis and degenerative musculoskeletal diseases. Gut Microbes, 18(1), 2661854. https://doi.org/10.1080/19490976.2026.2661854